Tutorial for Wannierizing Nb-Ti (mp-1216634). We will run VASP using atomate2, followed by a MLWF with Wannier90, but you could run VASP “by hand” if you’re not comfortable with atomate2.
Regardless, make sure the folder structure is the same as the one stated here.
Overview
In this tutorial you will run the following
| Step | Task |
|---|---|
| 1 | SCF calculation in atomate2 |
| 2 | Bandstructure (line mode) calculation in atomate2 |
| 3 | Bandstructure (uniform) calculation in atomate2 |
| 4 | Wannierization with SCDM using the result form Step 3, in atomate2 |
| 5 | Wannierization with LOCPROJ directly in the terminal |
| 6 | MLWF post optimization of the generated wannier functions |
Requirements
| Package | Version |
|---|---|
| atomate2 | 0.1.4 |
| jobflow | 0.3.1 |
| pymatgen | 2026.4.16 |
from atomate2.vasp.jobs.core import StaticMaker, NonSCFMaker
from atomate2.vasp.powerups import update_user_incar_settings, update_user_kpoints_settings
from jobflow.core.flow import Flow
from jobflow.managers.local import run_locally
from pymatgen.core import Structure, Lattice
from pymatgen.io.vasp import Kpoints
a = 2.84
b = 2.84
c = 4.65
structure = Structure.from_spacegroup(
"Cmmm",
Lattice.orthorhombic(a,b,c),
species=["Nb", "Ti"],
coords=[ [0, 0, 0], [1/2, 1/2, 1/2] ]
).get_primitive_structure()
static_job = StaticMaker(name=f"static").make(structure)
line_job = \
NonSCFMaker(name=f"line").make(
structure = static_job.output.structure,
prev_dir = static_job.output.dir_name,
)
uniform_job = \
NonSCFMaker(name=f"bands").make(
structure = static_job.output.structure,
prev_dir = static_job.output.dir_name,
)
scdm_job = \
NonSCFMaker(name=f"scdm").make(
structure = uniform_job.output.structure,
prev_dir = uniform_job.output.dir_name,
)
locproj_job = \
NonSCFMaker(name=f"locproj").make(
structure = uniform_job.output.structure,
prev_dir = uniform_job.output.dir_name,
)
Naively, one would proceed and run the computation. However, Wannierization works best when symmetry is turned off (helps with the gauge smoothness).
NSCF INCAR updates
| Parameter | Value | Reason |
|---|---|---|
ISYM | -1 | Symmetry might introduce extra gauge discontinuities |
ALGO | Normal | |
LWAVE | TRUE | Basis for Wannierizaton, will need to diagonalize $\hat{H}_{\rm KS}$ again |
LOCPROJ INCAR updates
| Parameter | Value | Reason |
|---|---|---|
LOCPROJ | “1 2 : s s p d : Hy” | Whichever projections we want to build orbitals from, see below for an explanation |
NUM_WANN | IGNORED | NUM_WANN is determined by the number of projectors in LOCPROJ |
ALGO | Eigenval or None | We just need to recalculate the energies or post-process using the current orbitals |
SCDM INCAR updates
| Parameter | Value | Reason |
|---|---|---|
LSCDM | TRUE | Turns on SCDM |
LWANNIER90 | TRUE | Turns on Wannier90 |
LWRITE_MMN_AMN | TRUE | Writes .mmn and .amn files for wannier90.x inputs |
ALGO | Eigenval or None | We just need to recalculate the energies or post-process using the current orbitals |
CUTOFF_MU | 23.0 | SCDM cutoff position, value specific to this example (but can be automated). NOT w.r.t. Fermi energy |
CUTOFF_SIGMA | 0.5 | SCDM cuttoff width |
NUM_WANN | 20 | Number of Wannier functions (or bands) |
The wannier90.amn and wannier90.mmn files contain the matrices $A_{mn}=\langle \psi_{n}| g_m\rangle $ and $M_{mn}=\langle \psi_{n}| \psi_{m}\rangle$ respectively. They are used as inputs for the MLWF procedure. The functions $g_m$ are initial projections in the LOCPROJ tag, or the SCDM orbitals.
Note that for SCDM, we are not bound by NUM_WANN, but for LOCPROJ we are bound by how many projectors we choose (e.g. a set of $s$ and $p$ orbitals have $1+3$ total bands in a non-spin polarized calculation).
For this examle, we choose $s$, $s$, $p$, $d$ to sum to 10 bands per element ($1+1+3+5$).
uniform_incar = {"ISYM": -1, "LWAVE": True, "NBANDS": 40}
scdm_incar = {"ALGO": "None", "ISTART": 2, "LWANNIER90": True, "LWRITE_MMN_AMN": True, "NUM_WANN": 20, "LSCDM": True, "CUTOFF_MU": 23.0, "CUTOFF_SIGMA": 0.5}
static_kpts = Kpoints.gamma_automatic((2,2,2))
uniform_kpts = Kpoints.gamma_automatic((6,6,6))
# update INCAR
uniform_job = update_user_incar_settings(uniform_job, uniform_incar)
scdm_job = update_user_incar_settings(scdm_job, scdm_incar)
# update kpoints for runs
static_job = update_user_kpoints_settings(static_job , static_kpts)
line_job = update_user_kpoints_settings(line_job, {"line_density": 10})
uniform_job = update_user_kpoints_settings(uniform_job, uniform_kpts)
scdm_job = update_user_kpoints_settings(scdm_job, uniform_kpts) # not really used as ALGO = None
flow = Flow([static_job, line_job, uniform_job, scdm_job], name="full_flow")
Notes on LOCPROJ
The local orbitals are defined as sites : angular character : radial character, and so 1 2 : s s p d: Hy means “project onto two sets of $s$, one set of $p$ and $d$ Hydrogenic orbitals for both elements in sites 1 and 2 of the POSCAR”.
The VASP section on LOCPROJ has more information about the types of orbital projections one can use.
Alternatively, for projections one can create a wannier90.win file beforehand with a projections block (similar to Q.E.)
begin projections
Ti : s;s;p;d;
Nb: s;s;p;d
end projections
However, I would not recommend this as VASP does not support hybrid projections in this format (e.g. the $sp-2$ projection will work when specified through LOCPROJ = 1 : sp-2 : Hy, but not through the wannier90.win file!)
Interpolating with Wannier90
Insert the following in the wannier90.win file that was created automatically by VASP. We will use the default gauge (i.e. no MLWF optimization)
num_iter = 0 # Don't optimize the spread
dis_num_iter = 0 # Don't do any disentanglement
write_hr = .true. # writes TB hamiltonian data
write_xyz = .true. # writes atomic positions and Wannier centres in cartesian coordinates
bands_plot = .true.
bands_num_points = 200
# kpath must match the one by VASP
begin kpoint_path
G 0.0 0.0 0.0 X 0.0 0.5 0.0
X 0.0 0.5 0.0 M 0.5 0.5 0.0
M 0.5 0.5 0.0 G 0.0 0.0 0.0
G 0.0 0.0 0.0 Z 0.0 0.0 0.5
Z 0.0 0.0 0.5 R 0.0 0.5 0.5
R 0.0 0.5 0.5 A 0.5 0.5 0.5
A 0.5 0.5 0.5 Z 0.0 0.0 0.5
R 0.0 0.5 0.5 X 0.0 0.5 0.0
M 0.5 0.5 0.0 A 0.5 0.5 0.5
end kpoint_path
Please rename the folders below to the corresponding output directories in your machine
from pymatgen.io.vasp import Vasprun
from pymatgen.electronic_structure.plotter import BSPlotter
from pymatgen.electronic_structure.bandstructure import BandStructure, BandStructureSymmLine, Kpoint
from pymatgen.electronic_structure.core import Spin
import numpy as np
import pandas as pd
from atomate2_wannier.plot_utils import make_pretty
LINE_PATH = "./line"
LOCPROJ_PATH = "./locproj"
SCDM_PATH = "./scdm"
vr = Vasprun(LINE_PATH + "/vasprun.xml.gz")
nscf = Vasprun(SCDM_PATH + "/vasprun.xml.gz")
bs = vr.get_band_structure(line_mode=True)
scdm_bands_raw = np.loadtxt(SCDM_PATH + "/wannier90_band.dat")
scdm_kpts_raw = np.loadtxt(SCDM_PATH + "/wannier90_band.kpt", skiprows=1)
scdm_ticks = pd.read_csv(SCDM_PATH + "/wannier90_band.labelinfo.dat", names=["label", "id", "dist", "x", "y", "z"], sep='\\s+')
scdm_ticks.loc[scdm_ticks.label == "G", "label"] = r"$\Gamma$"
locproj_bands_raw = np.loadtxt(LOCPROJ_PATH + "/wannier90_band.dat")
locproj_kpts_raw = np.loadtxt(LOCPROJ_PATH + "/wannier90_band.kpt", skiprows=1)
locproj_ticks = pd.read_csv(LOCPROJ_PATH + "/wannier90_band.labelinfo.dat", names=["label", "id", "dist", "x", "y", "z"], sep='\\s+')
num_wann_scdm = 20
num_wann_locproj = 20
num_kpts = scdm_kpts_raw.shape[0]
scdm_kpts = scdm_bands_raw[:, 0].reshape(num_wann_scdm, num_kpts)[0]
scdm_energies = scdm_bands_raw[:, 1].reshape(num_wann_scdm, num_kpts)
locproj_kpts = locproj_bands_raw[:, 0].reshape(num_wann_locproj, num_kpts)[0]
locproj_energies = locproj_bands_raw[:, 1].reshape(num_wann_locproj, num_kpts)
Show code
fig, ax = plt.subplots(2,1, dpi = 200, figsize=(4.0, 6), sharey=True)
make_pretty(fig,ax)
ax[0].plot(bs.distance, bs.bands[Spin.up].T - nscf.efermi, '.', color = 'C0', alpha=0.7, ms=3)
# ax[0].plot(scdm_kpts, scdm_energies.T - nscf.efermi, '-', color = 'k', lw = 1.5)
# ax[0].axhline(scdm_incar['CUTOFF_MU'] - nscf.efermi, ls='-.', color = 'r', lw = 2.0)
ax[0].set_xticks(scdm_ticks.dist)
ax[0].set_xticklabels(scdm_ticks.label)
[ax[0].axvline(x, lw=0.5, c='k') for x in scdm_ticks.dist]
ax[0].set_title("High symm. k-path")
ax[0].set_ylim(-60,40)
ax[0].set_xlim(scdm_ticks.dist.iloc[0], scdm_ticks.dist.iloc[-4])
ax[0].set_ylabel(r"$E-E_F$")
ax[1].plot(nscf.get_band_structure().bands[Spin.up].T - nscf.efermi, '.', color = 'C0', alpha=0.7, ms=3)
ax[1].set_xlabel("k-point index")
ax[1].set_xlim(0, nscf.get_band_structure().bands[Spin.up].shape[1])
ax[1].set_title("Uniform k-points")
# ax[1].plot(locproj_kpts, locproj_energies.T - nscf.efermi, '-', color = 'k', lw = 1.5)
# ax[1].set_xticks(scdm_ticks.dist)
# ax[1].set_xticklabels(scdm_ticks.label)
# [ax[1].axvline(x, lw=0.5, c='k') for x in scdm_ticks.dist]
# ax[1].set_title("LOCPROJ")
# ax[1].set_ylim(-60,40)
# ax[1].set_xlim(scdm_ticks.dist.iloc[0], scdm_ticks.dist.iloc[-4])
plt.tight_layout()
plt.show()
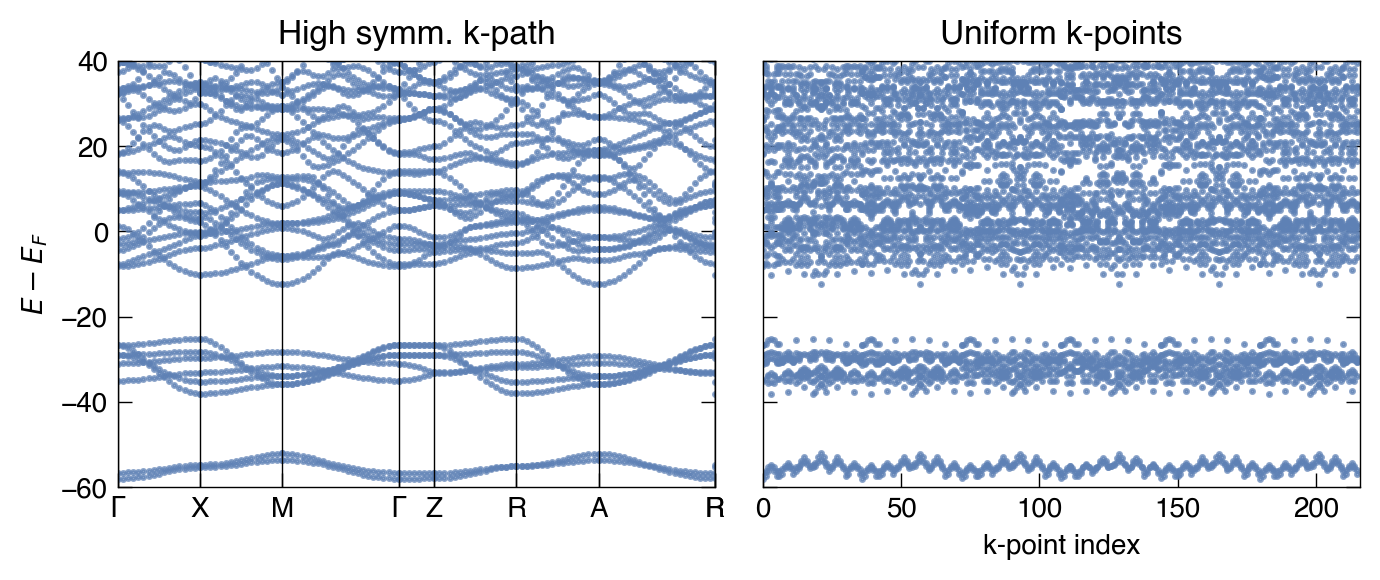
nscf.get_band_structure().bands[Spin.up].shape[1]
216
Show code
fig, ax = plt.subplots(2,1, dpi = 350, figsize=(5,8), sharex=True)
make_pretty(fig,ax)
ax[0].plot(bs.distance, bs.bands[Spin.up].T - nscf.efermi, '.', color = 'C0', alpha=0.7, ms=3)
ax[0].plot(scdm_kpts, scdm_energies.T - nscf.efermi, '-', color = 'k', lw = 1.5)
ax[0].axhline(scdm_incar['CUTOFF_MU'] - nscf.efermi, ls='-', color = 'r', lw = 1.0)
ax[0].set_xticks(scdm_ticks.dist)
ax[0].set_xticklabels(scdm_ticks.label)
[ax[0].axvline(x, lw=0.5, c='k') for x in scdm_ticks.dist]
ax[0].set_title("SCDM")
ax[0].set_ylim(-60,40)
ax[0].set_xlim(scdm_ticks.dist.iloc[0], scdm_ticks.dist.iloc[-4])
ax[0].set_ylabel(r"$E-E_F$")
ax[1].plot(bs.distance, bs.bands[Spin.up].T - nscf.efermi, '.', color = 'C0', alpha=0.7, ms=3)
ax[1].plot(locproj_kpts, locproj_energies.T - nscf.efermi, '-', color = 'k', lw = 1.5)
ax[1].set_xticks(scdm_ticks.dist)
ax[1].set_xticklabels(scdm_ticks.label)
[ax[1].axvline(x, lw=0.5, c='k') for x in scdm_ticks.dist]
ax[1].set_title("LOCPROJ")
ax[1].set_ylim(-60,40)
ax[1].set_ylabel(r"$E-E_F$")
ax[1].set_xlim(scdm_ticks.dist.iloc[0], scdm_ticks.dist.iloc[-4])
plt.tight_layout()
plt.show()
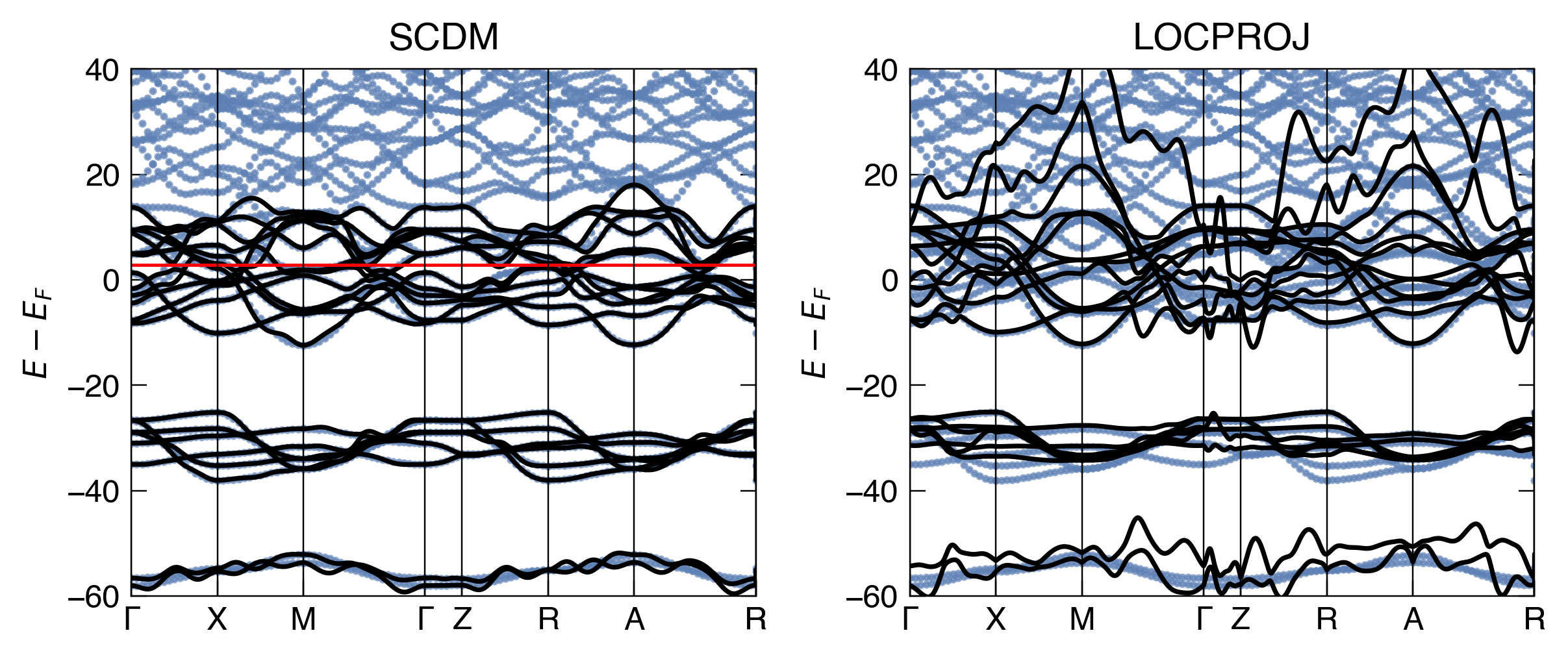
Note that SCDM gets a much better band-structure without any optimization (although we provided disentanglement options, whereas with LOCPROJ we did not disentangle, so not really an apples to apples comparison).
MLWF
The VASP2WANNIER90 can insert more information into the wannier90.win file through the WANNIER90_WIN tag. However, doing so requires multiline strings, which are not handled well by pymatgen (yet).
wannier90.win file before running VASP or else VASP will read this file and use the settings written in this file (e.g. when increasing k-points in the NSCF calculation, not removing the previous wannier90.win file will result in an error.)I detail here the parameters for MLWF generation through wannier90.x.
In WANNIER90_WIN tag or wannier90.win file
LOCPROJ
| Parameter | Value | Reason |
|---|---|---|
| num_iter | 1000 | Iterations for spread optimization |
| dis_num_iter | 500 | Need to optimize subspace before spread optimization (disentanglement) |
| dis_froz_min | 0 (eV) | Minimum of disentanglement window |
| dis_froz_max | 30 (eV) | Top of disentanglement window |
SCDM
| Parameter | Value | Reason |
|---|---|---|
| num_iter | 1000 | Iterations for spread optimization |
| dis_num_iter | 0 | Subspace is already disentangled, disentangling again can worsen the quality of the WFs |
Excluding bands
Usually, we are not interested in semi-core states, such as the ones below -50 eV. There is no tag in the INCAR for this, it is a parameter in the *.win file.
Unfortunately, for VASP<6.5.0 versions, the interface will not take into account the exclude_bands tag.
In the above example we have 2 semi-core states ($s$ orbitals) and six valence states ($p$ orbitals), so we exclude states 1-6 from the MLWF process.
In INCAR:
| Parameter | Value | Reason |
|---|---|---|
| WANNIER90_WIN | “exclude_bands = 6” | String that gets inserted in wannier90.win, how many bands to exclude from the overlap matrices |
SCDM_MLWF_PATH = "./scdm_mlwf"
LOCPROJ_MLWF_PATH = "./locproj_mlwf"
dis_froz_min = 0
dis_froz_max = 30
scdm_bands_mlwf = np.loadtxt(SCDM_MLWF_PATH + "/wannier90_band.dat")
scdm_mlwf_kpts = scdm_bands_mlwf[:, 0].reshape(num_wann_scdm, -1)[0]
scdm_mlwf = scdm_bands_mlwf[:, 1].reshape(num_wann_scdm, -1)
locproj_bands_mlwf = np.loadtxt(LOCPROJ_MLWF_PATH + "/wannier90_band.dat")
locproj_mlwf_kpts = locproj_bands_mlwf[:, 0].reshape(num_wann_locproj, -1)[0]
locproj_mlwf = locproj_bands_mlwf[:, 1].reshape(num_wann_locproj, -1)
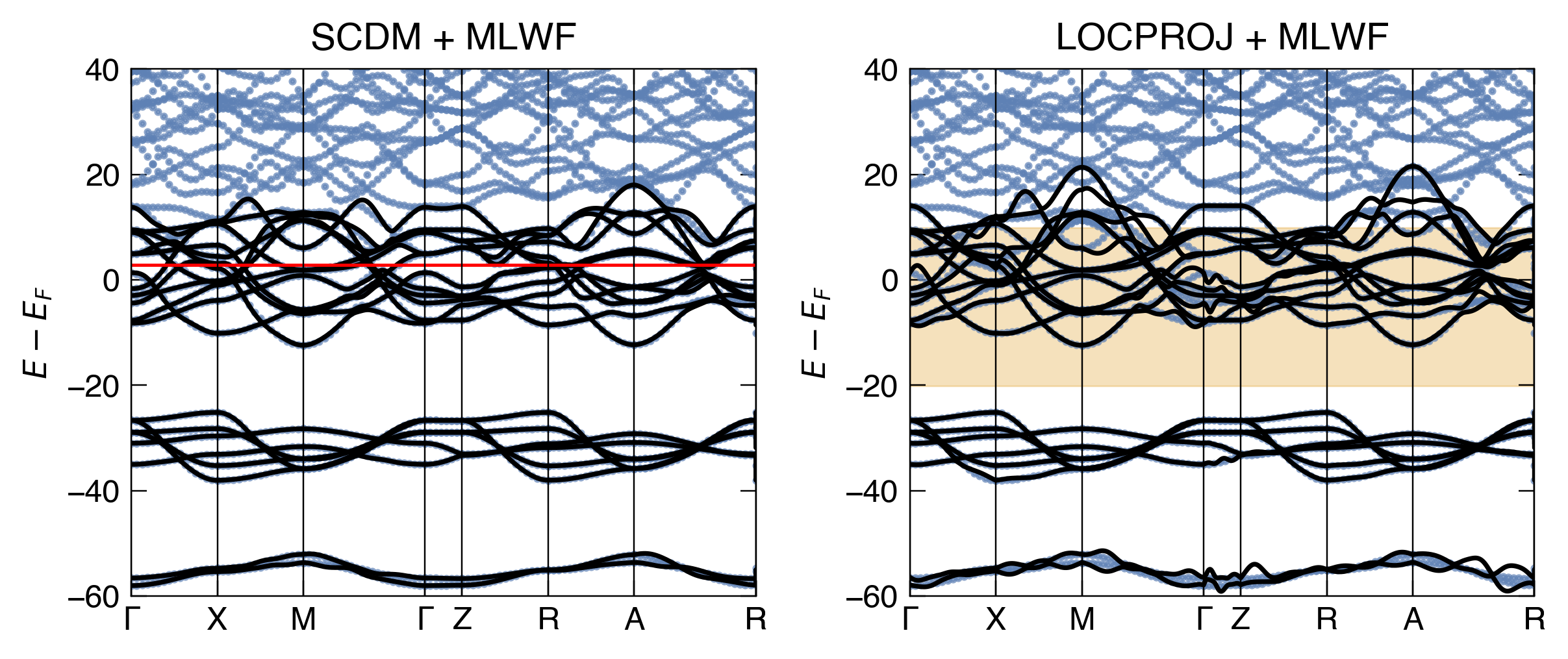
Show code
fig, ax = plt.subplots(2,1, dpi = 350, figsize=(5,8), sharex=True)
make_pretty(fig,ax)
ax[0].plot(bs.distance, bs.bands[Spin.up].T - nscf.efermi, '.', color = 'C0', alpha=0.7, ms=3)
ax[0].plot(scdm_mlwf_kpts, scdm_mlwf.T - nscf.efermi, '-', color = 'k', lw = 1.5)
ax[0].axhline(scdm_incar['CUTOFF_MU'] - nscf.efermi, ls='-', color = 'r', lw = 1)
ax[0].set_ylabel(r"$E-E_F$")
ax[1].plot(bs.distance, bs.bands[Spin.up].T - nscf.efermi, '.', color = 'C0', alpha=0.7, ms=3)
ax[1].plot(locproj_mlwf_kpts, locproj_mlwf.T - nscf.efermi, '-', color = 'k', lw = 1.5)
ax[1].fill_between(np.linspace(0,scdm_mlwf_kpts[-1],100), dis_froz_min - nscf.efermi, dis_froz_max - nscf.efermi, alpha=0.3, color='C1', label = "Frozen window")
ax[1].set_ylim(-60,40)
[ax[0].axvline(x, lw=0.5, c='k') for x in scdm_ticks.dist]
ax[0].set_title("SCDM + MLWF" )
[ax[1].axvline(x, lw=0.5, c='k') for x in scdm_ticks.dist]
ax[1].set_title("LOCPROJ + MLWF" )
ax[1].set_ylabel(r"$E-E_F$")
# limits
ax[1].set_xticks(scdm_ticks.dist)
ax[1].set_xticklabels(scdm_ticks.label)
ax[1].set_xlim(scdm_ticks.dist.iloc[0], scdm_ticks.dist.iloc[-4])
ax[0].set_ylim(-60,40)
# ax[1].legend(loc="upper right")
plt.tight_layout()
plt.show()
Show code
from atomate2_wannier import get_omega
from pathlib import Path
SCDM_MLWF_PATH = "./scdm_mlwf"
LOCPROJ_MLWF_PATH = "./locproj_mlwf"
scdm_omega = {s: get_omega(Path(SCDM_MLWF_PATH, "wannier90.wout")) for s in Spin}
proj_omega = {s: get_omega(Path(LOCPROJ_MLWF_PATH, "wannier90.wout")) for s in Spin}
# proj_omega = {s: get_omega(Path(dir, "proj", f"{elname}_{s.name}.wout")) for s in Spin}
fig, ax = plt.subplots(dpi = 250 , figsize=(4,2.4))
(fig, ax)
ax.plot(scdm_omega[Spin.up][:,-1], '-C0', label="SCDM")
# ax.plot(scdm_omega[Spin.down][:,-1], '-', label="PD+SCDM")
ax.plot(proj_omega[Spin.up][:,-1], '-C1', label="LOCPROJ")
ax.text(10**1, 90, "LOCPROJ", c="C1")
ax.text(10**.5, 55, "SCDM", c="C0")
xticks = [10^j for j in range(3)]
ax.axvline(500, lw=0.5, color='gray', ls="--")
ax.set_xticks(xticks)
# ax.plot(proj_omega[Spin.up][:,-1], '-C3', label="ED+AO")
ax.tick_params(axis='y', which='both', labelleft=True, labelright=True)
ax.set_ylabel(r"Sum of spreads $\Omega$ [$\AA^2$]")
ax.set_xlabel(r"Iterations")
ax.set_xscale('log')
ax.set_xlim(1, 1e3)
ax.set_ylim(10, 130)
# ax.legend()
plt.tight_layout()
plt.show()
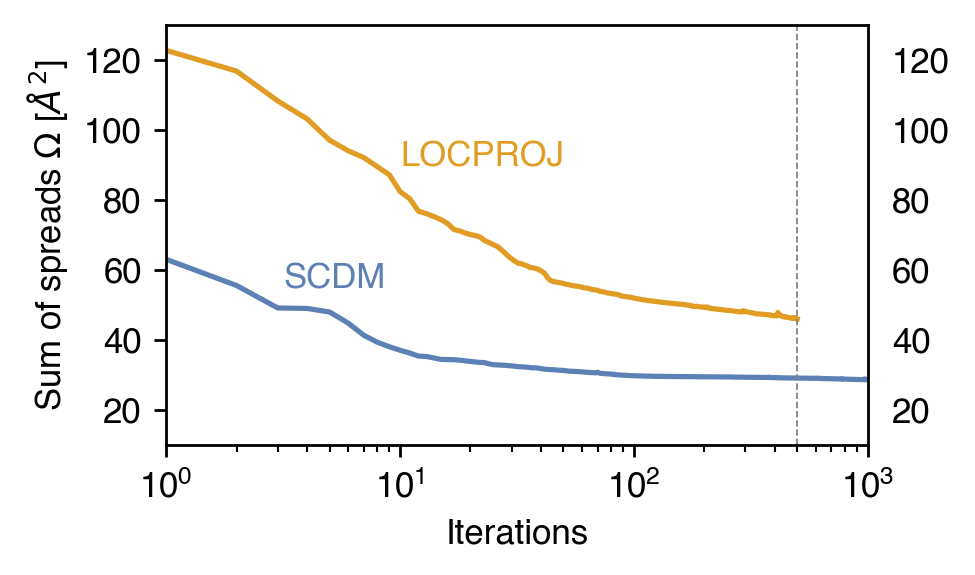
It seems the spread minimization of the LOCPROJ run did not reach finish optimizing, it would be best to increase the number of iterations.
from atomate2_wannier.utilities import parse_xyz
distances = []
for folder in [SCDM_MLWF_PATH, LOCPROJ_MLWF_PATH]:
closest = []
vasprun = Vasprun(Path(LOCPROJ_MLWF_PATH, "vasprun.xml"))
wann_centres = parse_xyz(Path(folder, "wannier90_centres.xyz"))[0]
sites = vasprun.final_structure.sites
dist_to_atom = {site.label: [] for site in sites}
struct = vasprun.final_structure
fracs = struct.lattice.get_fractional_coords(wann_centres)
dist_array = []
for (i,x0) in enumerate(fracs):
dist = {}
for site in struct.sites:
d, _ = site.lattice.get_distance_and_image(x0, site.frac_coords)
dist[site] = d
# get site with smallest distance to wannier center
closest_site = min(dist, key=dist.get)
# distances.append({closest_site: dist[closest_site]})
dist_array.append({closest_site.label: dist[closest_site]})
dist_to_atom[closest_site.label].append(dist[closest_site])
distances.append(dist_array)
Sanitized LOCPROJ: []
Sanitized LOCPROJ: []
/Users/ar/venvs/atomate2/lib/python3.12/site-packages/pymatgen/io/vasp/outputs.py:1353: UserWarning: No POTCAR file with matching TITEL fields was found in
if potcar := self.get_potcars(path):
/Users/ar/venvs/atomate2/lib/python3.12/site-packages/pymatgen/io/vasp/outputs.py:1364: UserWarning: No POTCAR file with matching TITEL fields was found in
potcar = self.get_potcars(path)
Atomic centered WFs
Even though SCDM has a great spread and can be very automatic compared to the LOCPROJ method, it fails in both symmetry and selective-localization. In other words, the orbitals generated will most definitely be far away from the atomic positions, and thus will not be very useful for tasks that require assigning wavefunctions to atoms (like estimating exchange interactions in TB2J).
Show code
fig, ax = plt.subplots(2,1, dpi = 350, figsize=(5,8), sharex=True)
make_pretty(fig,ax)
ax[0].axhline(0, lw=0.5, color="gray")
ax[1].axhline(0, lw=0.5, color="gray")
bins = np.linspace(0,4,10)
color_dict = {"Nb": "#61A57D", "Ti": "#78caff"}
lattice_const = struct.lattice.c
data = np.array([[[ val / c, color_dict[key]] for key,val in d.items()] for d in distances[0]], dtype=np.object_).squeeze()
[ax[0].plot(i, v[0], c=v[1], marker='o') for i,v in enumerate(data)]
data = np.array([[[ val / c, color_dict[key]] for key,val in d.items()] for d in distances[1]], dtype=np.object_).squeeze()
[ax[1].plot(i, v[0], c=v[1], marker='o') for i,v in enumerate(data)]
ax[1].set_xlabel("Wannier func. index")
# ax[1].set_xticks(range(0,21,5))
ax[1].set_ylabel(r"distance $ / a$")
ax[0].set_ylabel(r"distance $ / a$")
# ax[1].set_yticks([0,1])
ax[0].set_title("SCDM")
ax[1].set_title("LOCPROJ")
ax[1].set_yticks([0, 0.3])
ax[1].set_xticks(range(0,21, 10))
plt.tight_layout()
plt.show()
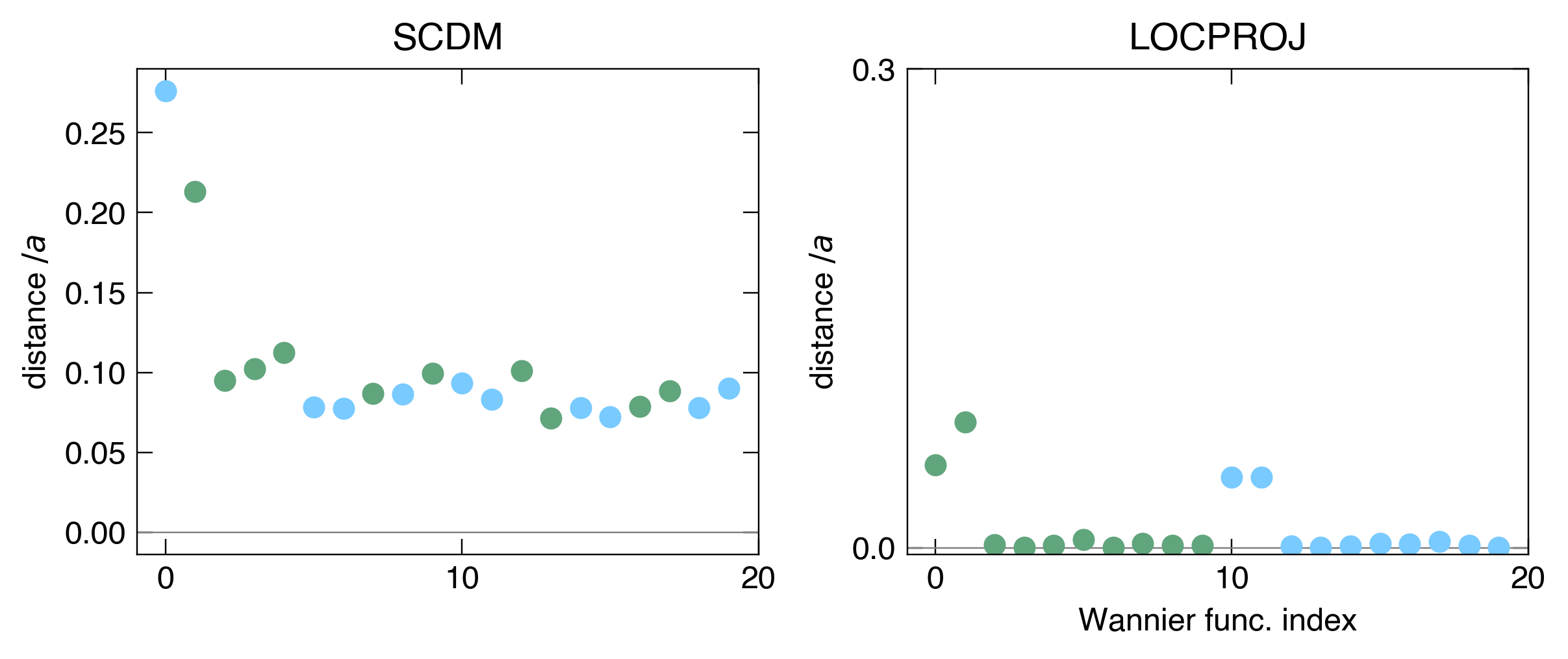
Interpolating the Hamiltonian ourselves (optional)
Wannier90 can interpolate the Hamiltonian in a path defined under kpoint_path. However, editing this file can become tedious. If we set write_hr and write_xyz to .true. in the wannier90.win file, then Wannier90 will write the Wannier Hamiltonian elements $\langle m\mathbf{0}| H |n\mathbf{R}\rangle$ and the cartesian lattice indices $\mathbf{R}$ needed to pefrorm the interpolation
$$ H_{mn}^W=\sum_{\mathbf{R}}e^{i\mathbf{k}\cdot\mathbf{R}}\langle m\mathbf{0}| H |n\mathbf{R}\rangle $$
We will perform an interpolation through $\Gamma \rightarrow X\rightarrow M \rightarrow \Gamma$
kp_sym = \
np.array([
[0.0, 0.0, 0.0], # G
[0.0, 0.5, 0.0], # X
[0.5, 0.5, 0.0], # M
[0.0, 0.0, 0.0], # G
])
num_sym = kp_sym.shape[0]
nk_tot = 2**8
kpath_to_interpolate = np.vstack([np.linspace(kp_sym[j], kp_sym[j+1], nk_tot // (num_sym - 1), endpoint=False) for j in range(num_sym - 1)])
k_distance = np.cumsum(np.linalg.norm(np.vstack([[0,0,0], np.diff(kpath_to_interpolate, axis=0)]), axis=1))
from TB2J.wannier import parse_atoms, parse_ham, parse_xyz
import scipy as sp
import gc
num_wann, hr, rvecs = parse_ham("./scdm_mlwf/wannier90_hr.dat")
xcart = parse_xyz("./scdm_mlwf/wannier90_centres.xyz")
atoms = parse_atoms("./scdm_mlwf/wannier90.win")
degen = rvecs.astype(float)
H_mnR_frac = {}
for R_frac, H in hr.items():
# R_frac is a tuple (i, j, k) in fractional coordinates
H_mnR_frac[R_frac] = H # Key is (i, j, k) fractional
R_vecs = np.array(list(H_mnR_frac.keys())) # fractional coordinates
H_vals = np.array(list(H_mnR_frac.values()))
H_vals_w = H_vals / degen[:, None, None] # unfold crystal symmetry
phases = np.exp(2j * np.pi * kpath_to_interpolate @ R_vecs.T)
H_mnK = (phases[:, :, None, None] * H_vals_w[None, :, :, :]).sum(axis=1)
H_mnK = 2*H_mnK # idk why we need twice its value to match dft, maybe a cartesian / fractional convention
# get eigenvalues
E_nk, U_mnk = sp.linalg.eigh(H_mnK)
gc.collect() # check if we can release some memory
59014
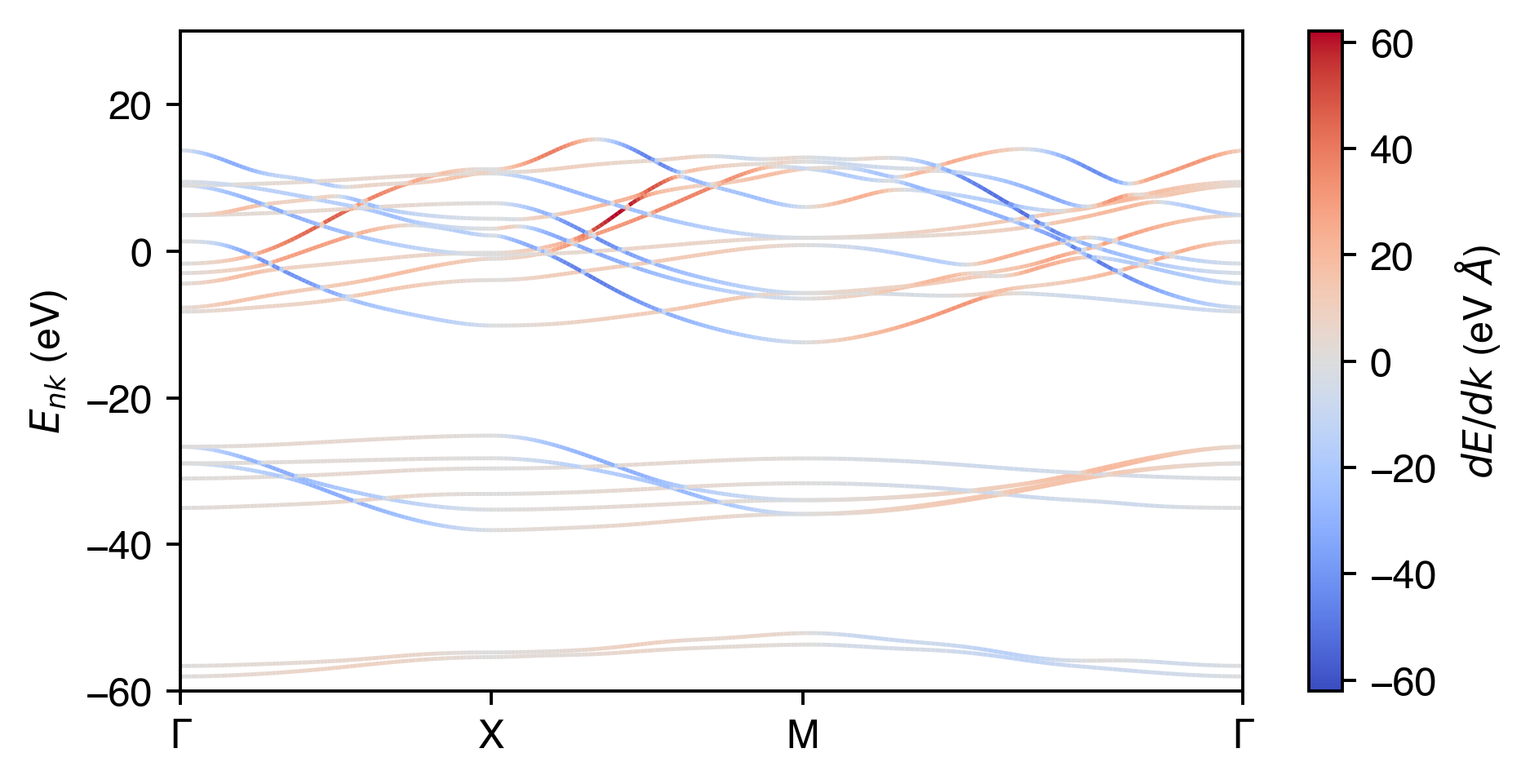
Calculating Bloch velocity
Show code
from matplotlib.collections import LineCollection
fig, ax = plt.subplots(dpi=350, figsize=(6,3))
band_velocities = np.array([np.gradient(band, k_distance) for band in E_nk.T])
vmin = np.min(band_velocities)
vmax = np.max(band_velocities)
vval = np.max([np.abs(vmin), vmax])
norm = plt.Normalize(vmin= -vval, vmax=vval)
for band, scalar_velocity in zip(E_nk.T, band_velocities):
points = np.array([k_distance / k_distance[-1], band - nscf.efermi]).T.reshape(-1, 1, 2)
segments = np.concatenate([points[:-1], points[1:]], axis=1)
# Pass the norm object to LineCollection
lc = LineCollection(segments, cmap='coolwarm', norm=norm, lw=1)
# Set the values used for colormapping
lc.set_array(scalar_velocity)
ax.add_collection(lc)
ax.set_xticks(scdm_ticks.dist / scdm_ticks.dist.iloc[3])
ax.set_xticklabels(scdm_ticks.label)
ax.set_xlim(0, 1)
ax.set_ylim(-60, 30)
ax.set_ylabel(r"$E_{nk}$ (eV)")
cbar = fig.colorbar(lc, ax=ax, label=r'$dE/dk$ (eV $\AA$)')
plt.show()